Click here to download this post
A 50 year-old female was evaluated for progressive symptoms of fainting, dizziness, and significant drop in blood pressure upon standing over the last six weeks.
She had abdominal discomfort, constipation, dry eyes or dry mouth (which may indicate Sjögren’s Disease), and Anhydrosis (inability to sweat or lack of sweating).
She had urinary symptoms of frequency and could not tolerate bright lights. All of these symptoms were new. Her blood pressure dropped 45 points from sitting to standing.
She also has low-normal epinephrine levels at rest when tested in the laboratory. Her pupils were dilated. She had no abnormal sensory or muscle abnormalities.
In the Autonomic Neuropathy laboratory, she showed evidence of impaired Sympathetic and Parasympathetic parameters.
Her heart rate response to deep breathing was impaired as was her Valsalva response indicating abnormalities of her cardiovagal system. Beat-to-beat blood pressure responses during Valsalva showed an absent overshoot, indicating Sympathetic abnormalities.
Because of the acute or subacute onset of symptoms in a middle-aged individual, autoimmune Autonomic Neuropathy was suspected.
Various autoimmune antibody tests were conducted, inducing antibodies to reflect Sjögren’s Disease (antibodies to SSA and SSB), Paraneoplastic antibodies (e.g., anti-Hu), and antibodies against Acetylcholine receptors were all negative. The patient began treatment with conventional medicines to treat Orthostatic Hypotension, including low-dose Midodrine (2.5 mg bid) and Mestinon (30 mg bid). While the orthostatic blood pressure was better controlled in time, other symptoms of constipation, dilated pupils, bright light sensitivity, and Hypo- or Anhydrosis, continued. The patient asked if she would benefit from a course of Prednisone or immunomodulating agents such as Intravenous Immunoglobulin (IVIG), as she had been reading up on the Internet, but she may still have an autoimmune type of Peripheral Autonomic Neuropathy that was not picked up by conventional autoantibody testing.
Orthostatic Hypotension is one form of autonomic dysfunction and one of the earliest, and perhaps most debilitating symptoms of autonomic neuropathy. Orthostatic Hypotension is also one form of Orthostatic Intolerance.
Orthostatic Hypotension presents as a significantly abnormal drop in blood pressure in response to upright posture, including standing or head-up tilt table test.
In fact any blood pressure response to standing that is less than a 10 mmHg increase in systolic blood pressure upon standing is considered abnormal.
Specifically, Orthostatic Hypotension is defined as a decrease in blood pressure upon standing of more than 20/10 mmHg pressure, and other change of less than a 10 mmHg increase in systolic blood pressure upon standing is considered to be Orthostatic Intolerance.
Other autonomic forms of Orthostatic Dysfunction include Postural Orthostatic Tachycardia Syndrome (an excessive increase in heart rate upon standing) and, rarely, Orthostatic Hypertension (an excessive increase in blood pressure upon standing).
While there are several underlying reasons for Orthostatic Dysfunction, other than autonomic dysfunction (e.g., venous valve dysfunction and dysfunction of the smooth muscles in the walls of the lower vasculature), the underlying autonomic dysfunction is known as Sympathetic Withdrawal.
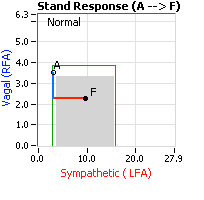
Normally, upon standing, the Parasympathetic first decrease to potentiate and minimize the (alpha-) Sympathetic response. The Parasympathetic 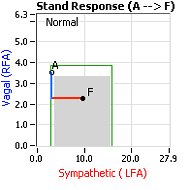 decrease is represented by the blue line decreasing, going down, in the figure, above, right. This begins the process of vasoconstriction to move blood up to the abdomen to help the heart pump blood to the brain. Then the Sympathetics increase (represented by the red line increasing, going to the right, in the figure, above right). This Sympathetic increase sustains the vasoconstriction and continues to shift the majority of the blood volume from the feet, against gravity, to the abdomen so that the heart may more easily pump it to the brain (see figure, above, right). Think of a car as the model. The Parasympathetics are the brakes and the Sympathetics are the accelerator. When stopped at a red light with your foot on the brakes and the light turns green, what is the first thing you do?
decrease is represented by the blue line decreasing, going down, in the figure, above, right. This begins the process of vasoconstriction to move blood up to the abdomen to help the heart pump blood to the brain. Then the Sympathetics increase (represented by the red line increasing, going to the right, in the figure, above right). This Sympathetic increase sustains the vasoconstriction and continues to shift the majority of the blood volume from the feet, against gravity, to the abdomen so that the heart may more easily pump it to the brain (see figure, above, right). Think of a car as the model. The Parasympathetics are the brakes and the Sympathetics are the accelerator. When stopped at a red light with your foot on the brakes and the light turns green, what is the first thing you do?
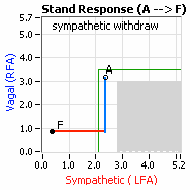 … You take your foot off the brakes. Even before you touch the accelerator, you begin to roll, you already begin to accelerate. Taking your foot off the brakes minimizes the amount gas (read that as Adrenaline) and acceleration (read that as Sympathetic stress) you need to reach your desired speed. The Parasympathetic and Sympathetic nervous systems normally act in much the same manner: first the Parasympathetics decrease to facilitate and minimize the Sympathetic response, and then the Sympathetics increase. Sympathetic Withdrawal is the abnormal decrease in alpha-Sympathetic activity upon standing (see figure, left).
… You take your foot off the brakes. Even before you touch the accelerator, you begin to roll, you already begin to accelerate. Taking your foot off the brakes minimizes the amount gas (read that as Adrenaline) and acceleration (read that as Sympathetic stress) you need to reach your desired speed. The Parasympathetic and Sympathetic nervous systems normally act in much the same manner: first the Parasympathetics decrease to facilitate and minimize the Sympathetic response, and then the Sympathetics increase. Sympathetic Withdrawal is the abnormal decrease in alpha-Sympathetic activity upon standing (see figure, left).
Note, women tend towards Postural Orthostatic Tachycardia Syndrome. This is due to the fact that, on average, women are born with physically 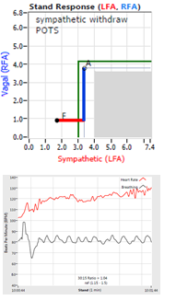 smaller hearts than men. Therefore, when their hearts become deconditioned, their hearts do not have the leverage to increase pressure to deliver more blood to the brain, so it resorts to the only other way and that is to increase rate to deliver more blood to the brain. This increased rate is Tachycardia (see figure, lower, right: the upper panel displays the Sympathetic Withdrawal and the lower panel displays the instantaneous respiratory (gray trace) and heart rate (red trace) during the first five-minutes of standing from a seated posture, note how the heart rate does not return to baseline as would be normal, but increases and continues to increase throughout the stand period and, for the most part, exceeds 120 bpm).
smaller hearts than men. Therefore, when their hearts become deconditioned, their hearts do not have the leverage to increase pressure to deliver more blood to the brain, so it resorts to the only other way and that is to increase rate to deliver more blood to the brain. This increased rate is Tachycardia (see figure, lower, right: the upper panel displays the Sympathetic Withdrawal and the lower panel displays the instantaneous respiratory (gray trace) and heart rate (red trace) during the first five-minutes of standing from a seated posture, note how the heart rate does not return to baseline as would be normal, but increases and continues to increase throughout the stand period and, for the most part, exceeds 120 bpm).
In all patients with Orthostatic Dysfunction, a deconditioned heart is a primary symptom. A deconditioned heart does not necessarily mean that the skeletal muscles of the body are deconditioned.
Patients with Orthostatic Dysfunction and deconditioned hearts are often in good physical condition and are (or were) able to exercise, even rigorously. In fact the exercise made them feel better (temporarily) because it used the skeletal muscles to help bring blood to the heart to improve circulation.
Their feet were warmer and, in less pain, and their brains were better perfused and more “awake.”
The exercise was a form of temporary, self-medication. While exercise is ultimately the best medicine to re-condition the heart, the alpha-Sympathetic nerves need to be “retrained” to respond properly and increase to cause the required vasoconstriction needed to support the heart.
Often this exercise needs to be low and slow, so as to not over-stress the nervous system. A standard to consider is 40 minutes of exercise per day, walking at no more than 2 mph, every day for six months.
On another note, Autonomic Dysfunction may involve multiple dysfunctions. Often, Orthostatic Dysfunction (Sympathetic Withdrawal) may be 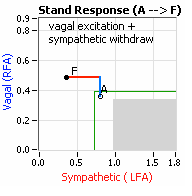 accompanied by a Vagal or Parasympathetic Excess (see figure, right). Parasympathetic Excess may be associated with Vasovagal Syncope. The Parasympathetic Excess (represented by the blue line increasing in the figure, right) is the Vagal component, followed by the Sympathetic Withdrawal. With Parasympathetic and Sympathetic Monitoring (P&S Monitoring, aka, Cardiorespiratory Monitoring) separate, but simultaneous measurements of Parasympathetic and Sympathetic nervous system activity is available in an easy to administer and perform test in the clinic. With documentation of both Sympathetic Withdrawal and Parasympathetic Excess, both conditions may be treated simultaneously: one treatment to reverse Sympathetic Withdrawal (e.g., Midodrine, Mestinon, or Alpha-Lipoic Acid) and one treatment to relieve Parasympathetic Excess (e.g., very, low-dose Anticholinergics or low and slow Exercise).
accompanied by a Vagal or Parasympathetic Excess (see figure, right). Parasympathetic Excess may be associated with Vasovagal Syncope. The Parasympathetic Excess (represented by the blue line increasing in the figure, right) is the Vagal component, followed by the Sympathetic Withdrawal. With Parasympathetic and Sympathetic Monitoring (P&S Monitoring, aka, Cardiorespiratory Monitoring) separate, but simultaneous measurements of Parasympathetic and Sympathetic nervous system activity is available in an easy to administer and perform test in the clinic. With documentation of both Sympathetic Withdrawal and Parasympathetic Excess, both conditions may be treated simultaneously: one treatment to reverse Sympathetic Withdrawal (e.g., Midodrine, Mestinon, or Alpha-Lipoic Acid) and one treatment to relieve Parasympathetic Excess (e.g., very, low-dose Anticholinergics or low and slow Exercise).
These are specific, common examples of Autonomic Neuropathy. For Autoimmune Autonomic Ganglionopathy (AAG) and Autoimmune Autonomic Neuropathy we need a deeper understanding of Autonomic Neuropathy and its causes.
An autoimmune mechanism where patients produce antibodies against neuronal tissue receptors is only one cause of Autonomic Neuropathy.
Furthermore, given that the Parasympathetic nervous system controls and coordinates the Immune system, recent evidence indicates that Parasympathetic Excess may induce autoimmunity through an excessively active immune system.
Autonomic Neuropathy is a malfunction of the Autonomic Nervous System (ANS) and is also referred to as Dysautonomia.
Generally, Autonomic Neuropathy refers to the peripheral involvement of the ANS involving the Parasympathetic and Sympathetic and Enteric Nervous Systems, which are all parts of the ANS, and, specifically, the Enteric Nervous System is considered to be a part of Parasympathetic Nervous System.
There are cases of autonomic dysfunction which affect the brain or spinal cord, such as Multiple System Atrophy, but these are separate from Peripheral Autonomic Neuropathies.
Because the ANS controls or coordinates all organs and systems of the body, all organs and systems are affected, some perhaps more so than others; at least at first.
Therefore, patients with broader or more advanced autonomic neuropathies may have urinary symptoms (such as urinary retention or urinary incontinence), gastrointestinal symptoms (such as abdominal pain, nausea, gastroparesis, diarrhea, constipation or swallowing difficulties), and may have disturbances of heart rate where the heart rate can be very fast, very slow, or have swings in between.
Patients may also have significant drops in blood pressure, a condition known as orthostatic hypotension, especially when stranding from a lying or sitting position.
Many patients have exercise intolerance and cannot increase their heart rate effectively when they exert themselves.
They can have abnormal pupil responses or sweat disturbances: either sweating too much or too little.
Patients may have dry eyes or dry mouth (so called Sicca Syndrome, aka. Sjögren’s Disease). The patients may also fail to recognize, or have defective, warning symptoms of hypoglycemia.
Most importantly, people with Peripheral Autonomic Neuropathies should have no evidence of Parkinson’s disease or abnormalities of the cerebellum with gait disturbances as is seen in more serious diseases known as Multi-System Atrophy (MSA).
When a person presents with symptoms of Peripheral Autonomic Neuropathy, we often seek the cause. Many have had antecedent, recent viral or bacterial infections.
Some may have had concussions or head trauma or a motor vehicle accident. Occasionally, we see people with severe, acutely emotional stress.
Patients with Ehlers-Danlos Syndrome (EDS) or Hypermobility usually develop a more gradual type of autonomic dysfunction and not an acute or subacute type.
Diabetes is probably the most common cause of autonomic dysfunction and also causes gradual nerve damage throughout the body.
We can also see certain medicines, such as use of cancer chemotherapy or radiation therapy causing injury to nerves which can produce autonomic neuropathies.
A rare disease, Amyloidosis (AL) which affects organs in the nervous system due to build up to abnormal proteins can occur, specifically those related to light chains or a familial type related to a different type of abnormal protein called Transthyretin (hATTR).
The latter is a build-up of a genetic mutation that results in a misfolded Transthyretin protein. This causes Amyloid deposits in various organs, including the heart, nerves and GI tract.
When it occurs in the nerves, patients can develop Autonomic Neuropathy and Orthostatic Hypotension.
Neurodegenerative disease, including Parkinson’s disease or Lewy Body Dementia and even Multiple Sclerosis, eventually lead to autonomic dysfunction.
Interestingly, although Parkinson’s disease and Lewy Body Dementia affect the central nervous system, the autonomic dysfunction that results is due to a Peripheral Autonomic Neuropathy. There are certain hereditary causes of Autonomic Neuropathy.
Some autoimmune diseases, however, can cause autonomic neuropathies.
This is when a person’s body produces antibodies that attack nervous system components. One such case is Autoimmune Autonomic Ganglionopathy.
Occasionally, similar mechanisms are seen in people who have cancer where they produce antibodies against their nerve tissue that can affect the Peripheral, the Sensory-Motor, and Central nervous systems in these people.
This is known as a Paraneoplastic Syndrome. We can send out for testing of antibodies if this is suspected.
Other autoimmune diseases, in which the immune system damages nerve fibers, include Sjögren’s syndrome, Systemic Lupus Erythematosus, Rheumatoid Arthritis, Mixed Collagen Vascular Diseases, Celiac Disease, and occasionally Guillain-Barre Syndrome.
Chronic Alcoholism can also cause chronic Peripheral Autonomic Neuropathy. Although rare, Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) can have some elements of autonomic dysfunction.
Usually autoimmune diseases can come on quickly, such Guillain-Barre Syndrome, in which autoantibodies attack the nervous system. At times, they can occur subacutely and rarely chronically evolve.
Eloquent rabbit and other animal experiments have shown that Autoimmune Autonomic Neuropathy may be caused by autoantibodies that the body produces against nerve tissue.
A human study[1] followed 112 patients with type 1 Diabetes and upon examination found the presence of circulating antibody to ANS structures.
They concluded that circulating antibody to autonomic structures was associated with development of autonomic dysfunction in young diabetic patients. They found this to be independent of blood sugar control.
Their perspective study demonstrated that the detection of circulating autoantibodies in the nervous system and subsequently over time the development of autonomic dysfunction most likely having a cause-and-effect relationship.
In this study, they also tested for somatic neuropathy with deep tendon reflexes, ankle reflexes, and vibratory perception to follow the evolution of sensory types of neuropathy found in diabetics.
Blood sugar control when it was poor appeared to accelerate into sensory neuropathy abnormalities that were followed with these physical examination parameters, but blood sugar did not predict the Peripheral Autonomic Neuropathy manifestations of the autoimmune components.
Autoantibodies to ANS tissues preceded the development of Autonomic Neuropathy in many of these patients. Type 1 Diabetic patients who developed Cardiac Autonomic Neuropathy had a prevalence of 68% antibody positivity when tested, which was significantly higher compared to antibody-negative patients.
The most impaired test was Parasympathetic Nervous System response to deep breathing, which is mainly mediated by the Parasympathetic Nervous System.
It is believed that autoimmune mechanisms that target Sympathetic and Parasympathetic structures play a significant causative role in the development and progression of autonomic dysfunction in type 1 diabetics, long-term, and the finding of autoantibodies in the blood, even in type 1 diabetics who do not have Autonomic Neuropathy predicts, with high positive predictive value, those who will develop Autonomic Neuropathy.
Autonomic Neuropathy is a continuum, starting with Peripheral Autonomic Neuropathy and ultimately progressing to and ending with Cardiovascular Autonomic Neuropathy (CAN). From P&S Monitoring, Peripheral Autonomic Neuropathy is characterized by abnormal challenge responses (that is to deep breathing, Valsalva, or stand or tilt) with normal resting responses. The next phase of Autonomic Neuropathy is Diabetic Autonomic Neuropathy (DAN, if the patient is diagnosed with Diabetes) or Advanced Autonomic Dysfunction (AAD). DAN or AAD are characterized by abnormally low, resting Parasympathetic or abnormally low, resting Sympathetic activity, but the resting Parasympathetic activity is greater than 0.1 bpm2 (see figure, right). One branch activity low is sufficient for AAD, both indicates a more advanced AAD. AAD or DAN is associated with more overt symptoms of Autonomic Neuropathy, and significantly greater morbidity risk leading to numbers of co-morbidity. Unfortunately, the co-morbidities tend to be treated independently, leading to significantly increased numbers of medications, rather than seeking the underlying cause and treating that to relieve multiple symptoms and co-morbidities. While DAN or AAD is not life threatening, it does threaten quality of life.
treating that to relieve multiple symptoms and co-morbidities. While DAN or AAD is not life threatening, it does threaten quality of life.
End stage Autonomic Neuropathy, CAN, is defined by resting Parasympathetic activity less than 0.1 bpm2 (see figure, right), regardless of the level of resting Sympathetic activity or challenge responses. Returning to the car analogy, this would be like worn brakes. Regardless of the state of the accelerator, without any brakes, you may not stop and the possible crash may be life threatening. It is similar with CAN. Without significant levels of resting Parasympathetic activity to balance resting Sympathetic activity, mortality risk escalates, and the risk is stratified by the level of imbalance between the P&S branches, known as Sympathovagal Balance (SB: for CAN patients, the range of normal SB is 0.4 < SB < 1.0). Normalizing SB, treats CAN, and normalizes mortality risk.
Other studies have shown relationships between autoantibodies and development of autonomic dysfunction.
These have shown an independent relationship with blood sugar control as well. The mechanism in autoimmunity in type 1 diabetics is similar to what is seen in Paraneoplastic dysautonomias in which patients with cancers develop antibodies against their Acetylcholine receptors and develop severe autonomic dysfunction.
The higher the levels of antibodies, the worse the autonomic dysfunction is in these patients.
This indicates a therapeutic role for Acetylcholine inhibitors in the improvement in autonomic dysfunction. It is interesting that type 1 diabetics also have an autoimmune mechanism where there is an active B-cell response against pancreatic and nervous system tissue. It may well be that autoantibodies attack both the pancreas and the ANS.
The mechanisms differentiating sensory neuropathy and Autonomic Neuropathy in type 1 Diabetes are different.
The sensory neuropathy is associated with blood sugar control. The Autoimmune Autonomic Neuropathy is not.
Also, 30% of patients who develop signs of peripheral somatic neuropathy, such as sensory or motor abnormalities, do not have associated autonomic dysfunction.
There appears to be two different mechanisms operating:
(1) sensory neuropathy in diabetes appears to be effected by poor blood sugar control and may be related to metabolic or oxidative end products with poorly controlled diabetes; whereas, (2) the diabetic type 1 Autonomic Neuropathy appears to be autoimmune as an individual produces antibodies against neuronal tissue and is not related to the blood sugar level. The authors stated that they do not know whether the autoantibodies enhanced the presentation of antigens or a lead to Channelopathies. Therefore, based on results of animal experimental studies and the perspective followup of over 16 years of type 1 diabetes, it is now established that autoantibodies may cause a Peripheral Autonomic Neuropathy.
Autoimmune Autonomic Neuropathy appears to affect the Acetylcholine Ganglionic receptors. It is an antibody-mediated response that usually presents with autonomic failure involving the Sympathetic, Parasympathetic and Enteric nervous system.
Various portions of the Acetylcholine receptor can be affected by antibodies attacking different locations within the receptor.
Usually, this evolves over acute or subacute course. 50% of individuals will have antibodies to the Acetylcholine receptor and the other half will not.
However, the half that do not have antibodies detected and do not have any Paraneoplastic antibodies detected probably still have unknown antibodies for which we have not been able to search.
Higher titers of antibodies usually correlate with the severity of the Dysautonomia. Patients with high antibody titers in a study by Vernino in the Annals of Neurology, 2003, had a combination of Sicca Syndrome with marked dry eyes and dry mouth, abnormal pupillary light response, upper gastrointestinal symptoms and neurogenic bladder.
Higher antibody titers appear to be associated with more frequent Cholinergic Dysautonomia. Chronic cases occasionally occur and are difficult to separate from advanced autonomic failure, which is a separate disorder, quite rare, which can remain chronic or evolve into a more severe central disorder or a degenerative disorder, such as Parkinson’s or MSA.
Orthostatic Hypotension, widespread Hypo- or Anhidrosis, dry mouth, dry eyes, sexual dysfunction, urinary retention, impaired pupillary responses, reduced heart rate variability and gastrointestinal symptoms ranging from gastroparesis to postprandial abdominal pain, to diarrhea and more commonly constipation can occur.
Rarely, intestinal pseudo-obstruction, a severe form of hypomotility of the GI tract can occur. Oftentimes, a virus, a recent immunization, or surgical procedure is reported prior to onset of symptoms which are similar to what we see with Guillain-Barre Syndrome, which does not usually involves the autonomics, or only mildly, but involves the sensory and motor components of the nervous system.
Interestingly, in the treatment of advanced Autoimmune Autonomic Neuropathy, if one has high levels of anti-Acetylcholine antibodies, they will come down.
Also, high levels of antibodies against Acetylcholine receptors are associated more with acute and subacute onset and more severe Dysautonomia with prominent Cholinergic features (i.e., Sicca complex, prominent gastrointestinal dysmotility and pupillary abnormalities).
Low titers are often seen in more indolent and chronic phenotypes. As mentioned, half of patients may not even have titers that are positive for antibodies and a yet unidentified antibody may be the culprit.
Occasionally, in the chronic forms that evolve patients present with Orthostatic Hypotension as the more prominent feature and oftentimes they cannot stand for periods of time and may even faint.
Low plasma Catecholamine levels, such as reduced Norepinephrine release, are seen in patients with autoimmune widespread dysautonomia. Sudomotor testing, which reflects postganglionic dysfunction indicating dysautonomia, is easily performed in laboratories and clinics.
Studies of Sympathetic cardiac innervation with MIBG scans showing abnormal cardiac uptake in Norepinephrine spillover tests may confirm a postganglionic dysfunction. It is important to differentiate between acute and subacute onset Pandysautonomias with prominent Cholinergic abnormalities, as these respond well to immunotherapy, such as IVIG, Prednisone or other immune suppressive agents.
If only one feature of dysautonomia is present, usually antibody titers to Acetylcholine receptors are not present. An individual could have an isolated entity known as Chronic Idiopathic Anhidrosis. These patients have heat intolerance.
They have a better prognosis as this is a restrictive type Dysautonomia. However, only about 16% of people test positive for Acetylcholine receptors with this disorder, and they usually have a low titer.
The Burning Feet Syndrome, usually due to Small Fiber Neuropathy seen often in diabetics, usually affects small unmyelinated nerve fibers, but some may not have any etiology, and it is postulated that this could be an autoimmune mechanism with distal fiber neuropathies. However, these patients have low positivity of Acetylcholine receptors.
Chronic Pseudointestinal Obstruction, where patients get frequent obstruction of the bowel, a severe dysmotility disorder may be caused by many mechanisms. No specific antigen or antibodies have been identified.
However, if one has positive antibodies against the Acetylcholine receptor, this may represent a form of Autoimmune Autonomic Neuropathy affecting the GI tract more selectively. In other words, this could be another variant of Autoimmune Autonomic Neuropathy caused by Autoimmune Autonomic Ganglionopathy (AAG).
Remember, seronegativity or absence of antibody responses, measured in patients with acute and subacute and occasionally chronic peripheral autonomic neuropathies does not exclude an autoimmune mechanism.
It just may imply that the responsible autoantibody has not yet been identified. Some of these patients will respond to steroids and immunosuppressive agents such as IVIG and it is worthwhile considering this. Sandroni and Low in a paper, Other Autonomic Neuropathies Associated with Ganglionic Antibody Production, concluded that “similar phenotypes may have very different pathogenetic mechanisms” and “idiopathic” should not equate “autoimmune.”
While AAG patients do not typically have sensory abnormalities, some may describe minor sensory symptoms such as tingling, but with objective testing, sensory loss is not present, however, they have preserved reflex knee jerks, tickle sensation and so forth.
Immunomodulator therapy, such as Prednisone, IVIG, and other immunosuppressive agents may be very useful when used early in patients with Autoimmune Autonomic Neuropathy.
The higher the titers, for example, greater than 1 mmol/spot per liter, usually implies that one can improve with therapies.
Also, the more severe Orthostatic Hypotension patients with high levels of Acetylcholine receptor antibodies appear to improve with immunomodulator therapy. Both seropositive and seronegative AAG patients may respond to therapies, including plasma exchange and some combinations of immunosuppressive therapy especially if they do not respond to IVIG initially.
The clinical features of AAG reflect impairment of Sympathetic function with Orthostatic Hypotension, Syncope, Anhidrosis, Parasympathetic dysfunction (including, dry mouth, dry eyes, and impaired pupillary constriction), and Enteric dysfunction (including, gastrointestinal dysmotility, constipation, gastroparesis and rarely pseudo-obstruction)[2].
In regard to the Enteric Nervous System, there were two main plexus, the Myenteric (Auerbach’s) and Submucosal (Meissner’s neurons).
The Enteric Nervous System controls most gut functions, such as secretion, absorption, vascular tone and motility.
An enteric ganglionitis is an inflammatory neuropathy with inflammation and immunological insult to the intrinsic innervation supplying the GI tract. It may be associated with Paraneoplastic Syndrome and even infections such as Chagas Disease.
There are diffuse lymphoid infiltrates in the small intestine, and this can cause pseudo-obstruction or infiltration of myenteric ganglia and can also cause Achalasia, which is a contraction and motility disorder of the Esophagus.
Autoantibodies, including antineuronal antibodies, are associated with this disorder, and it is oftentimes associated also with Paraneoplastic or cancer syndromes. Clinical features of Enteric Ganglionitis include, dysmotility and delayed transit depending on what is affected in the gastrointestinal tract, whether it be the Esophagus, lower esophageal sphincter, stomach with gastroparesis ,or colon with an intestinal pseudo-obstruction and colonic inertia and even megacolon.
Paraneoplastic syndromes can cause a Peripheral Autonomic Neuropathy even before cancer becomes manifested. Oftentimes, they present as a subacute sensory neuropathy. These patients may usually have a small cell cancer and anti-Hu antibodies.
As mentioned earlier, other types of neuropathy, such as the sensorimotor neuropathy, Guillain-Barre Disease, and Chronic Inflammatory Demyelinating Polyneuropathy (CIDP), Brachial Plexopathy and Vasculitis Neuropathy may cause autonomic dysfunction in addition to sensory symptoms and sensory ataxia. Oftentimes, some of the sensory impairments are painful. Fiber loss is predominate in small myelinated and unmyelinated fibers. These can have similar antibodies detected as is seen in Paraneoplastic syndromes.
Connective tissue diseases can be associated with subacute neuropathies. This has been seen frequently with primary Sjogren’s syndrome where seven forms of neuropathy can be identified. A variable degree of autonomic dysfunction occurs with these collagen vascular and connective tissue diseases.
They may have Hypo-or Anhidrosis, abdominal pain, constipation, and diarrhea. These mechanisms may be different than autoimmune type autoantibodies seen in the conventional AAG patients. In these instances, T cells attack tissue or ischemia due to vasculitis may be operative. Interestingly, in many of these collagen vascular connective tissue vascular dysautonomias, SSA and SSB antibodies, which are often seen in Sjogren’s syndrome normally are not present.
In addition to Guillain-Barre, subclinical autonomic dysfunction has been reported in up to 25% of CIDP patients involving both Parasympathetic and Sympathetic components. Vasomotor and Sudomotor fibers are involved when the Sympathetic systems is affected. Autoimmune antibodies may not be present in these syndromes.
Alexander Szali , [Autoimmune Diseases, 2013] discussed autonomic involvement in subacute and chronic immune mediating neuropathies. He concluded that autonomic function may be impaired in subacute and chronic immune mediated neuropathies in which Sympathetic, Parasympathetic and Enteric arms of the ANS are affected.
When a physician sees Orthostatic Hypotension, gastrointestinal dysmotility, pseudo-obstruction, urinary retention, etc., one should be alerted to the fact that this could be an autoimmune mechanism. Also, one should be alert for the possibility of underlying occult cancer when an Autonomic Paraneoplastic disorder is suspected.
In an editorial by Muppidi, February 2018, in Clinical Autonomic Research, the author writes that Ganglionic Acetylcholine Receptor Antibodies are known to have a pathological role in AAG as an individual can produce antibodies against the Ganglionic Nicotinic Acetylcholine Receptor and disrupt cholinergic transmission at the Sympathetic and Parasympathetic ganglia.
This is the mechanism behind the Pandysautonomia. One should have a low threshold for ordering ganglionic AChR antibodies in patients with acute and subacute onset focal or generalized autonomic dysfunction syndromes.
Muppidi makes a distinction between those that are seropositive and have positive antibody levels, and those who have negative antibody levels. Those with negative antibody levels, or seronegative patients, appear to respond to high dose steroids whereas those who have positive autoantibody responses appear to more respond to plasma chains, IVIG or Rituximab.
The author postulates that there may be different underlying mechanisms in patients who have seropositive and seronegative AAG, and they propose a cell-mediated or inflammatory immune process rather than antibody-related mediated mechanism in those patients who are seronegative who may respond to high dose steroids.
Different assays test for Nicotinic Acetylcholine receptors. Conventionally, Radioimmunoprecipitation (RIP) assays have been used for sensitive detection of autoantibodies to Ganglionic Acetylcholine Receptors in serum of patients with AAG. In Japan, they have developed a Luciferase Immunoprecipitation System (LPS) which does not involve radionuclide administration. As mentioned earlier, one can do a cardiac MIBG scan which will show decreased cardiac uptake, which also can be seen in Lewy Body Disease and Parkinson’s Disease as well as Dementia with Lewy Bodies in these Peripheral Autonomic Neuropathies.
The heart-to-mediastinum ratio is calculated and if low in these patients the ratio reflects a peripheral mechanism of autonomic dysfunction.
AAG should not be confused with Myasthenia Gravis (MG) in which there is an Autoimmune Channelopathy that is caused by autoantibodies to the neuromuscular junction apparatus. In 80%, of these patients, these autoantibodies are noted against the muscle-type of Nicotinic Acetylcholine Receptor, not the ganglionic-type as seen in AAG.
High levels of antibodies in AAG patients are seen in patients with more significant autonomic dysfunction.
However, Ganglionic Anticholinergic Antibodies have been found in patients with Postural Orthostatic Tachycardia Syndromes only. Chronic Idiopathic Pseudo-Obstruction patients typically have chronic idiopathic Anhidrosis and Distal Small Fiber Neuropathy albeit in low titers as we have previously discussed.
Interestingly, several researches have also reported that patients with other neuroimmunological disorders, such as Myasthenia Gravis, Lambert-Eaton Myasthenic Syndrome, Guillain-Barre Syndrome, and Chronic Inflammatory Demyelinating Polyneuropathy may have antibodies to ganglionic Acetylcholine receptors and autonomic symptoms.
In 2009, researchers reporting in the Journal of Immunotherapy Cancer, describe a seronegative AAG from dual immune checkpoint inhibition in patients with Metastatic Melanoma.
This is a very sophisticated new class of cancer treating agent using Immune Checkpoint Inhibitor therapy.
It described a patient who developed symptoms of nausea, constipation, weight loss, fatigue and hypotension with systolic blood pressures as low as 70 and holding the Immune Checkpoint Inhibition caused resolution of the symptoms. In these patients, antibodies against Anticholinergic Receptors, anti-GAD 65 antibodies, Paraneoplastic Syndrome Antibodies (Mayo Clinic panel), ANA, Lyme, Syphilis and HIV testing were all negative. The patient also responded to treatment with pulse doses of IV Solumedrol and received IVIG.
In summary, AAG is one form of an autoimmune autonomic dysfunction syndrome due to autoantibodies. When it is seropositive with high antibody titers, autonomic dysfunction is usually quite severe, and we can follow antibody titers which lower with treatment.
They respond more to immunosuppressive agents such as IVIG. It appears that seronegative patients with features consistent with AAG respond better to steroids, and this may reflect a cell-mediated and not a humoral mechanism.
Patients with autonomic neuropathy often have Orthostatic Intolerance, severe GI symptoms with nausea, vomiting, early satiety, constipation, bloating and may even present with Achalasia and Paralytic Ileus. Sudomotor dysfunction in these patients is abnormal as there can be postganglionic disorders.
Pupillary dysfunction with bilateral Mydriasis, which reflects Parasympathetic denervation, is often prominently seen in AAG. We refer to this as an Adie Pupil. However, some cases of pupil dysfunction can be mixed problems with Sympathetic and Parasympathetic dysfunction.
There is also a slow form of AAG which resembles another disorder, Pure Autonomic Failure, which is more of a neurodegenerative disease due to an Alpha Synucleinopathy disorder.
In regard to our clinical vignette, which we presented at the beginning of this treatise, this patient appears to have a seronegative type of Autoimmune Autonomic Neuropathy. Consideration for immunotherapy and immunomodulating therapy should be given although some literature suggests that high-dose steroids may be a better first option.
While not the most common cause of Peripheral Autonomic Neuropathy, Autoimmune Autonomic Neuropathy does exist and one needs to think of it, test for it and follow the clinical course clearly to be able to make the diagnosis and initiate early treatment.
For more information visit our autonomic dysfunction center.
About the Author
Nicholas L. DePace, MD, FACC is a board-certified cardiologist and Medical Director of Franklin Cardiovascular Associates. A graduate of the Mount Sinai School of Medicine, Dr. DePace has decades of clinical, academic, and research experience and has held faculty appointments as a Clinical Professor of Medicine, becoming one of the youngest full professors in Philadelphia at the time of his appointment.
Dr. DePace specializes in the diagnosis and treatment of autonomic nervous system dysfunction (dysautonomia), including POTS, autonomic dysfunction associated with Ehlers-Danlos syndrome (EDS), chronic fatigue, and anxiety-like conditions that are frequently misdiagnosed. He is nationally recognized for his work on parasympathetic and sympathetic (P&S) nervous system imbalance, a core mechanism underlying many complex chronic disorders.
In addition to treating patients from across the United States, Dr. DePace is a prolific clinical researcher and author of multiple nationally distributed medical textbooks published by Springer and W.W. Norton, focusing on autonomic dysfunction, mitochondrial disorders, cardiovascular disease, and mind–body medicine.
 View Dr. DePace’s professional profile
View Dr. DePace’s professional profile
View medical books by Dr. DePace
[1] Maria Zanone MM, Raviolo A, Coppo E, Trento M, Trevisan M, Cavallo F, Favaro E, Passera P, Porta M, Camussi G. Association of Autoimmunity to Autonomic Nervous Structures With Nerve Function in Patients With Type 1 Diabetes: A 16-Year Prospective Study . Diabetes Care Apr 2014, 37 (4) 1108-1115; DOI: 10.2337/dc13-2274.
[2] Winston and Vernino, 2010, Current Opinions in Neurology


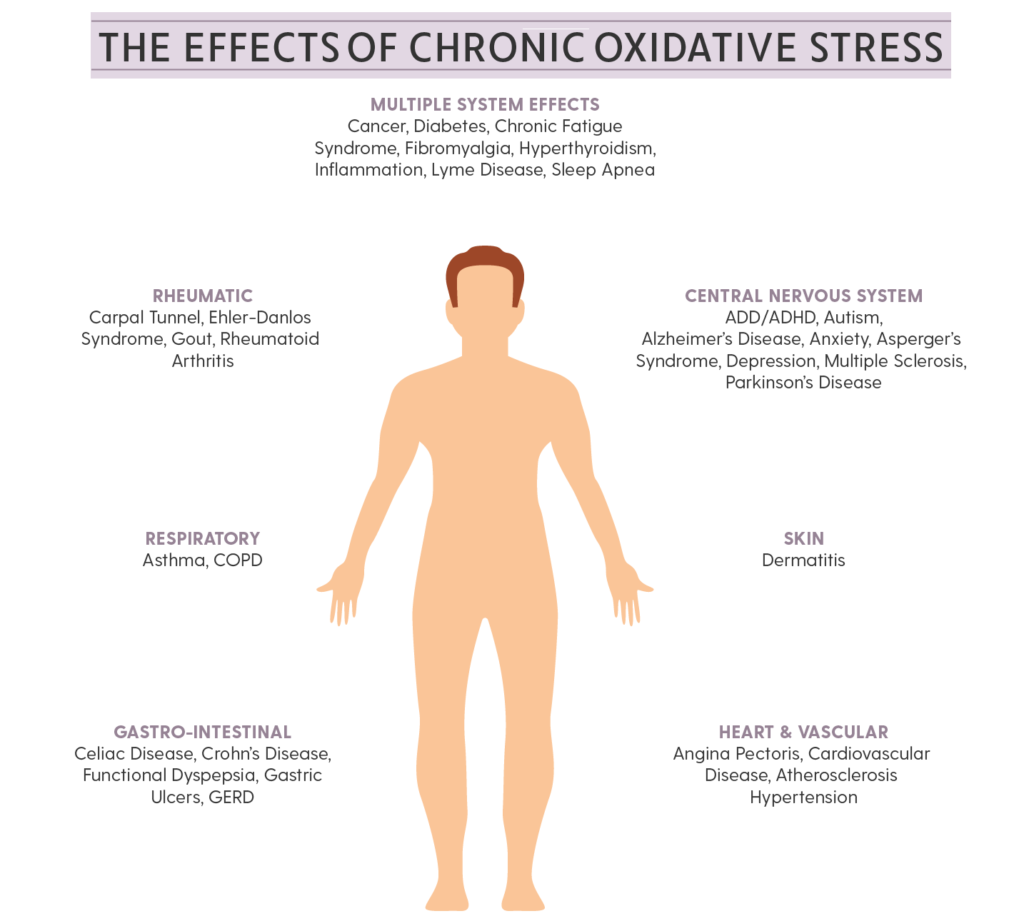
 waste (pollution) generated in the process of producing power. Mitochondria are the power plants of the cells and the body. It produces Adenosine Triphosphate (ATP) as the energy molecule, and some of the waste products (pollutants) are Free Radicals. Under healthy conditions, the body uses these Free Radicals to advantage, as mentioned above. Under unhealthy conditions, the body requires Antioxidants to neutralize the Free Radicals that are not used. Under chronic conditions, the body tends to need more than it is able to produce. In all of these conditions, there is an appropriate Antioxidant-Oxidant Balance that sustains health. While the exact amounts are unknown, fortunately there is no such things as too many Antioxidants. It is like having “too many” fire extinguishers. The less used the better and if they are never used, they are not wasted. To that end, a well-established and maintained pool of Antioxidants is always healthful.
waste (pollution) generated in the process of producing power. Mitochondria are the power plants of the cells and the body. It produces Adenosine Triphosphate (ATP) as the energy molecule, and some of the waste products (pollutants) are Free Radicals. Under healthy conditions, the body uses these Free Radicals to advantage, as mentioned above. Under unhealthy conditions, the body requires Antioxidants to neutralize the Free Radicals that are not used. Under chronic conditions, the body tends to need more than it is able to produce. In all of these conditions, there is an appropriate Antioxidant-Oxidant Balance that sustains health. While the exact amounts are unknown, fortunately there is no such things as too many Antioxidants. It is like having “too many” fire extinguishers. The less used the better and if they are never used, they are not wasted. To that end, a well-established and maintained pool of Antioxidants is always healthful. The body has natural Antioxidants to sequester, or neutralize, Free Radicals to prevent oxidative stress from injuring tissues and destroying cells. Natural Antioxidants include Vitamins A, C, & E, Glutathione, Selenium, Alpha Lipoic Acid (ALA) and Coenzyme Q-10 (CoQ10). Many scientists feel that ALA is the ideal antioxidant because it is both a lipid and water soluble (it can dissolve in both lipid and water environments) and can cross the Blood-Brain Barrier. It is absorbed rapidly through the Gastrointestinal (GI) tract high up in the digestive system and it is immediately available to neutralize free radicals quickly. It has also been shown to recycle Vitamin C and Vitamin E in the body. Vitamin C is only water soluble and Vitamin E is only lipid soluble. Because ALA is both liquid and lipid soluble, it can pass the Blood-Brain Barrier and increase available brain energy. Not only can ALA recycle Vitamin C & E but also Glutathione. Glutathione is probably the most important intracellular Antioxidant.
The body has natural Antioxidants to sequester, or neutralize, Free Radicals to prevent oxidative stress from injuring tissues and destroying cells. Natural Antioxidants include Vitamins A, C, & E, Glutathione, Selenium, Alpha Lipoic Acid (ALA) and Coenzyme Q-10 (CoQ10). Many scientists feel that ALA is the ideal antioxidant because it is both a lipid and water soluble (it can dissolve in both lipid and water environments) and can cross the Blood-Brain Barrier. It is absorbed rapidly through the Gastrointestinal (GI) tract high up in the digestive system and it is immediately available to neutralize free radicals quickly. It has also been shown to recycle Vitamin C and Vitamin E in the body. Vitamin C is only water soluble and Vitamin E is only lipid soluble. Because ALA is both liquid and lipid soluble, it can pass the Blood-Brain Barrier and increase available brain energy. Not only can ALA recycle Vitamin C & E but also Glutathione. Glutathione is probably the most important intracellular Antioxidant.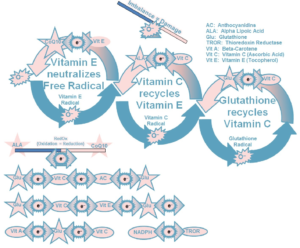 Antioxidant and is synthesized within the Mitochondria and consists of three Amino Acids, Cysteine, Glutamic Acid, and Glycine. Glutathione is not easily absorbed orally and cannot pass through the Mitochondrial membrane so easily. Therefore, anything that preserves the body’s natural production of Glutathione and keeps the concentration up is valuable. This is where ALA comes in as a very important Antioxidant. It recycles Glutathione and replenishes the body’s stores. Glutathione is a very important component of several enzyme systems in the body that are organ-protective from disease. There is some data to suggest that ALA is also an excellent chelating agent and protects us from heavy metals, although this is beyond the scope of this discussion.
Antioxidant and is synthesized within the Mitochondria and consists of three Amino Acids, Cysteine, Glutamic Acid, and Glycine. Glutathione is not easily absorbed orally and cannot pass through the Mitochondrial membrane so easily. Therefore, anything that preserves the body’s natural production of Glutathione and keeps the concentration up is valuable. This is where ALA comes in as a very important Antioxidant. It recycles Glutathione and replenishes the body’s stores. Glutathione is a very important component of several enzyme systems in the body that are organ-protective from disease. There is some data to suggest that ALA is also an excellent chelating agent and protects us from heavy metals, although this is beyond the scope of this discussion.
 decrease is represented by the blue line decreasing, going down, in the figure to the right. This begins the process of vasoconstriction to move blood up to the abdomen to help the heart pump blood to the brain. Then the
decrease is represented by the blue line decreasing, going down, in the figure to the right. This begins the process of vasoconstriction to move blood up to the abdomen to help the heart pump blood to the brain. Then the 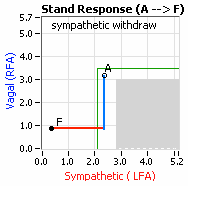 acceleration (read that as Sympathetic stress) you need to reach your desired speed. The Parasympathetic and Sympathetic nervous systems normally act in much the same manner: first the Parasympathetics decrease to facilitate and minimize the Sympathetic response, and then the Sympathetics increase. Sympathetic Withdrawal is the abnormal decrease in alpha-Sympathetic activity upon standing (see figure, left).
acceleration (read that as Sympathetic stress) you need to reach your desired speed. The Parasympathetic and Sympathetic nervous systems normally act in much the same manner: first the Parasympathetics decrease to facilitate and minimize the Sympathetic response, and then the Sympathetics increase. Sympathetic Withdrawal is the abnormal decrease in alpha-Sympathetic activity upon standing (see figure, left).
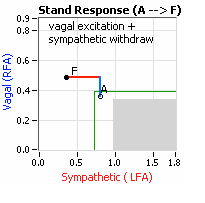 accompanied by a Vagal or Parasympathetic Excess (see figure, right). Parasympathetic Excess may be associated with Vasovagal Syncope. The Parasympathetic Excess (represented by the blue line increasing in the figure, right) is the Vagal component, followed by the Sympathetic Withdrawal. With Parasympathetic and Sympathetic Monitoring (P&S Monitoring, aka, Cardiorespiratory Monitoring) separate, but simultaneous measurements of Parasympathetic and Sympathetic nervous system activity is available in an easy to administer and perform test in the clinic. With documentation of both Sympathetic Withdrawal and Parasympathetic Excess, both conditions may be treated simultaneously: one
accompanied by a Vagal or Parasympathetic Excess (see figure, right). Parasympathetic Excess may be associated with Vasovagal Syncope. The Parasympathetic Excess (represented by the blue line increasing in the figure, right) is the Vagal component, followed by the Sympathetic Withdrawal. With Parasympathetic and Sympathetic Monitoring (P&S Monitoring, aka, Cardiorespiratory Monitoring) separate, but simultaneous measurements of Parasympathetic and Sympathetic nervous system activity is available in an easy to administer and perform test in the clinic. With documentation of both Sympathetic Withdrawal and Parasympathetic Excess, both conditions may be treated simultaneously: one