Click Here to Download this Blog Post – The “Soft Skeleton” Problem (Part 7)
By Dr. Nicholas L. DePace, M..D., F.A.C.C – Cardiologist specializing in autonomic dysfunction, Ehlers-Danlos syndrome and POTS.
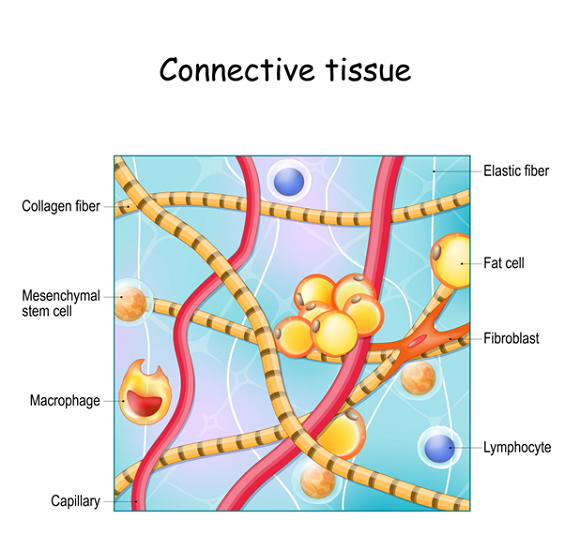
Heritable Connective Tissue Disorders (HCTDs) are caused by the body making collagen incorrectly.
- Collagen is the Body’s “Glue” Collagen is described as the body’s “soft skeleton.” It is found in every organ, blood vessel, and nerve. It gives your body its shape and holds your organs in place. When your body creates faulty collagen, it leads to disorders like Marfan syndrome, Stickler syndrome, and the most common one: Hypermobile Ehlers-Danlos Syndrome (hEDS).
- Ruling Out Danger It is critical for doctors to figure out exactly which disorder a patient has. While most patients have hEDS (which is painful but not fatal), there are rare types (like Vascular EDS) that can be life-threatening. Doctors need to rule those out first to ensure the patient is safe.
- The Name Matters Less Than the Treatment There is currently no genetic test for hEDS. Doctors often argue over labels—whether to call it hEDS, Hypermobility Spectrum Disorder (HSD), or the older term “Joint Hypermobility Syndrome.” The specific label doesn’t matter much because the treatment is the same: focus on relieving the pain, fatigue, and functional issues.
- The “Hallmark” Symptoms For most patients (especially young women), the biggest clue isn’t just flexible joints—it is Autonomic Dysfunction. This means the body’s automatic systems are out of sync. Common symptoms include:
- POTS: A racing heart when standing up.
- Chronic Fatigue: Feeling exhausted all the time.
- Thermoregulatory issues: Sweating too much or trouble controlling body temperature.
- Easy bruising.
In short, while flexible joints are a sign, the internal symptoms (like heart rate and fatigue) are often what bother the patient the most and lead to the diagnosis.
It is important to seek out a clinician with expertise in EDS to make an accurate diagnosis and create a treatment plan. One of the nation’s leading centers is Franklin Cardiovascular Associates, under the direction of Nicholas DePace, MD, FACC. They are located in Sicklerville, New Jersey. franklincardiovascular.com, (856) 589-6034
About the Author
Nicholas L. DePace, MD, FACC is a board-certified cardiologist and Medical Director of Franklin Cardiovascular Associates. A graduate of the Mount Sinai School of Medicine, Dr. DePace has decades of clinical, academic, and research experience and has held faculty appointments as a Clinical Professor of Medicine, becoming one of the youngest full professors in Philadelphia at the time of his appointment.
Dr. DePace specializes in the diagnosis and treatment of autonomic nervous system dysfunction (dysautonomia), including POTS, autonomic dysfunction associated with Ehlers-Danlos syndrome (EDS), chronic fatigue, and anxiety-like conditions that are frequently misdiagnosed. He is nationally recognized for his work on parasympathetic and sympathetic (P&S) nervous system imbalance, a core mechanism underlying many complex chronic disorders.
In addition to treating patients from across the United States, Dr. DePace is a prolific clinical researcher and author of multiple nationally distributed medical textbooks published by Springer and W.W. Norton, focusing on autonomic dysfunction, mitochondrial disorders, cardiovascular disease, and mind–body medicine.
👉 View Dr. DePace’s professional profile
👉 View medical books by Dr. DePace